

产品中心
当前位置:首页>产品中心Anti-Fibrinogen alpha chain
货号: bs-7548R 基本售价: 1380.0 元 规格: 100ul
- 规格:100ul
- 价格:1380.00元
- 规格:200ul
- 价格:2200.00元
产品信息
- 产品编号
- bs-7548R
- 英文名称
- Fibrinogen alpha chain
- 中文名称
- 纤维蛋白原A链抗体
- 别 名
- FGA; Fib2; FIBA_HUMAN; Fibrinogen alpha chain; fibrinogen alpha chain isoform alpha preproprotein; Fibrinogen alpha/alpha E chain [Precursor]; fibrinogen alpha chain isoform alpha-E preproprotein; fibrinogen alpha chain isoform alpha preproprotein.
- 规格价格
- 100ul/1380元购买 200ul/2200元购买 大包装/询价
- 说 明 书
- 100ul 200ul
- 研究领域
- 心血管
- 抗体来源
- Rabbit
- 克隆类型
- Polyclonal
- 交叉反应
- Human, Mouse, Rat, Dog, Pig, Cow, Horse, Rabbit,
- 产品应用
- WB=1:500-2000 ELISA=1:500-1000 IHC-P=1:400-800 IHC-F=1:400-800 IF=1:100-500 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
- 分 子 量
- 91kDa
- 细胞定位
- 分泌型蛋白
- 性 状
- Lyophilized or Liquid
- 浓 度
- 1mg/ml
- 免 疫 原
- KLH conjugated synthetic peptide derived from human Fibrinogen alpha chain:61-160/866
- 亚 型
- IgG
- 纯化方法
- affinity purified by Protein A
- 储 存 液
- 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
- 保存条件
- Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
- PubMed
- PubMed
- 产品介绍
- background:
The protein encoded by this gene is the alpha component of fibrinogen, a blood-borne glycoprotein comprised of three pairs of nonidentical polypeptide chains. Following vascular injury, fibrinogen is cleaved by thrombin to form fibrin which is the most abundant component of blood clots. In addition, various cleavage products of fibrinogen and fibrin regulate cell adhesion and spreading, display vasoconstrictor and chemotactic activities, and are mitogens for several cell types. Mutations in this gene lead to several disorders, including dysfibrinogenemia, hypofibrinogenemia, afibrinogenemia and renal amyloidosis. Alternative splicing results in two isoforms which vary in the carboxy-terminus. [provided by RefSeq, Jul 2008]
Function:
Fibrinogen has a double function: yielding monomers that polymerize into fibrin and acting as a cofactor in platelet aggregation.
Subunit:
Heterohexamer; disulfide linked. Contains 2 sets of 3 non-identical chains (alpha, beta and gamma). The 2 heterotrimers are in head to head conformation with the N-termini in a small central domain.
Subcellular Location:
Secreted.
Tissue Specificity:
Plasma.
Post-translational modifications:
The alpha chain is not glycosylated.
Forms F13A-mediated cross-links between a glutamine and the epsilon-amino group of a lysine residue, forming fibronectin-fibrinogen heteropolymers.
About one-third of the alpha chains in the molecules in blood were found to be phosphorylated.
Conversion of fibrinogen to fibrin is triggered by thrombin, which cleaves fibrinopeptides A and B from alpha and beta chains, and thus exposes the N-terminal polymerization sites responsible for the formation of the soft clot. The soft clot is converted into the hard clot by factor XIIIA which catalyzes the epsilon-(gamma-glutamyl)lysine cross-linking between gamma chains (stronger) and between alpha chains (weaker) of different monomers.
Phosphorylation sites are present in the extracellular medium.
DISEASE:
Defects in FGA are a cause of congenital afibrinogenemia (CAFBN) [MIM:202400]. This is a rare autosomal recessive disorder characterized by bleeding that varies from mild to severe and by complete absence or extremely low levels of plasma and platelet fibrinogen. Note=The majority of cases of afibrinogenemia are due to truncating mutations. Variations in position Arg-35 (the site of cleavage of fibrinopeptide a by thrombin) leads to alpha-dysfibrinogenemias.
Defects in FGA are a cause of amyloidosis type 8 (AMYL8) [MIM:105200]; also known as systemic non-neuropathic amyloidosis or Ostertag-type amyloidosis. AMYL8 is a hereditary generalized amyloidosis due to deposition of apolipoprotein A1, fibrinogen and lysozyme amyloids. Viscera are particularly affected. There is no involvement of the nervous system. Clinical features include renal amyloidosis resulting in nephrotic syndrome, arterial hypertension, hepatosplenomegaly, cholestasis, petechial skin rash.
Similarity:
Contains 1 fibrinogen C-terminal domain.
SWISS:
P02671
Gene ID:
2243
Database links:
Entrez Gene: 2243HumanEntrez Gene: 14161Mouse
Omim: 134820Human
SwissProt: P02671Human
SwissProt: Q99K47Mouse
Unigene: 351593Human
Unigene: 88793Mouse
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
- 产品图片
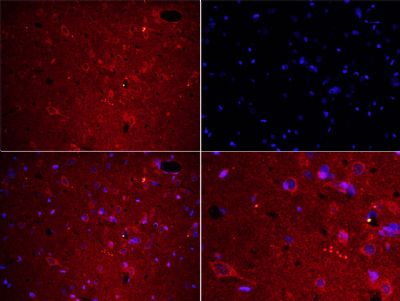 Tissue/cell: rat brain tissue;4% Paraformaldehyde-fixed and paraffin-embedded;
Tissue/cell: rat brain tissue;4% Paraformaldehyde-fixed and paraffin-embedded;
Antigen retrieval: citrate buffer ( 0.01M, pH 6.0 ), Boiling bathing for 15min; Blocking buffer (normal goat serum,C-0005) at 37℃ for 20 min;
Incubation: Anti-Fibrinogen alpha chain Polyclonal Antibody, Unconjugated(bs-7548R) 1:200, overnight at 4°C; The secondary antibody was Goat Anti-Rabbit IgG, Cy3 conjugated (bs-0295G-Cy3)used at 1:200 dilution for 40 minutes at 37°C. DAPI(5ug/ml,blue,C-0033) was used to stain the cell nuclei