

产品中心
当前位置:首页>产品中心Anti-CDMP1
货号: bs-6580R 基本售价: 1380.0 元 规格: 100ul
- 规格:100ul
- 价格:1380.00元
- 规格:200ul
- 价格:2200.00元
产品信息
- 产品编号
- bs-6580R
- 英文名称
- CDMP1
- 中文名称
- 软骨衍生形态发生蛋白1/GDF 5抗体
- 别 名
- Cartilage derived morphogenetic protein 1; Cartilage-derived morphogenetic protein 1; CDMP-1; CDMP1; GDF-5; Gdf 5; GDF5_HUMAN; Growth differentiation factor 5; Growth/differentiation factor 5; LAP4; Radotermin.
- 规格价格
- 100ul/1380元购买 200ul/2200元购买 大包装/询价
- 说 明 书
- 100ul 200ul
- 研究领域
- 心血管 细胞生物 信号转导 干细胞 生长因子和激素 转录调节因子
- 抗体来源
- Rabbit
- 克隆类型
- Polyclonal
- 交叉反应
- Human, Mouse, Rat, Dog, Pig, Cow, Horse, Rabbit,
- 产品应用
- WB=1:500-2000 ELISA=1:500-1000 (石蜡切片需做抗原修复)
not yet tested in other applications.
optimal dilutions/concentrations should be determined by the end user.
- 分 子 量
- 55kDa
- 细胞定位
- 细胞膜 分泌型蛋白
- 性 状
- Lyophilized or Liquid
- 浓 度
- 1mg/ml
- 免 疫 原
- KLH conjugated synthetic peptide derived from human CDMP1/GDF5:201-300/501
- 亚 型
- IgG
- 纯化方法
- affinity purified by Protein A
- 储 存 液
- 0.01M TBS(pH7.4) with 1% BSA, 0.03% Proclin300 and 50% Glycerol.
- 保存条件
- Store at -20 °C for one year. Avoid repeated freeze/thaw cycles. The lyophilized antibody is stable at room temperature for at least one month and for greater than a year when kept at -20°C. When reconstituted in sterile pH 7.4 0.01M PBS or diluent of antibody the antibody is stable for at least two weeks at 2-4 °C.
- PubMed
- PubMed
- 产品介绍
- background:
Defects in GDF5 are the cause of acromesomelic chondrodysplasia Grebe type (AMDG) . Acromesomelic chondrodysplasias are rare hereditary skeletal disorders characterized by short stature, very short limbs, and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMDG is an autosomal recessive form characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet.Defects in GDF5 are the cause of acromesomelic chondrodysplasia Hunter-Thompson type (AMDH). AMDH is an autosomal recessive form of dwarfism. Patients have limb abnormalities, with the middle and distal segments being most affected and the lower limbs more affected than the upper. AMDH is characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet.Defects in GDF5 are the cause of brachydactyly type C (BDC). BDC is an autosomal dominant disorder characterized by an abnormal shortness of the fingers and toes.
Function:
Could be involved in bone and cartilage formation. Chondrogenic signaling is mediated by the high-affinity receptor BMPR1B.
Subunit:
Homodimer; disulfide-linked (By similarity).
Subcellular Location:
Secreted.
Tissue Specificity:
Predominantly expressed in long bones during embryonic development.
DISEASE:
Defects in GDF5 are the cause of acromesomelic chondrodysplasia Grebe type (AMDG) [MIM:200700]. Acromesomelic chondrodysplasias are rare hereditary skeletal disorders characterized by short stature, very short limbs, and hand/foot malformations. The severity of limb abnormalities increases from proximal to distal with profoundly affected hands and feet showing brachydactyly and/or rudimentary fingers (knob-like fingers). AMDG is an autosomal recessive form characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet.
Defects in GDF5 are the cause of acromesomelic chondrodysplasia Hunter-Thompson type (AMDH) [MIM:201250]. AMDH is an autosomal recessive form of dwarfism. Patients have limb abnormalities, with the middle and distal segments being most affected and the lower limbs more affected than the upper. AMDH is characterized by normal axial skeletons and missing or fused skeletal elements within the hands and feet.
Defects in GDF5 are the cause of brachydactyly type C (BDC) [MIM:113100]. BDC is an autosomal dominant disorder characterized by an abnormal shortness of the fingers and toes.
Defects in GDF5 are the cause of Du Pan syndrome (DPS) [MIM:228900]; also known as fibular hypoplasia and complex brachydactyly. Du Pan syndrome is a rare autosomal recessive condition characterized by absence of the fibulae and severe acromesomelic limb shortening with small, non-functional toes. Although milder, the phenotype resembles the autosomal recessive Hunter-Thompson and Grebe types of acromesomelic chondrodysplasia.
Defects in GDF5 are a cause of symphalangism proximal syndrome (SYM1) [MIM:185800]. SYM1 is characterized by the hereditary absence of the proximal interphalangeal (PIP) joints (Cushing symphalangism). Severity of PIP joint involvement diminishes towards the radial side. Distal interphalangeal joints are less frequently involved and metacarpophalangeal joints are rarely affected whereas carpal bone malformation and fusion are common. In the lower extremities, tarsal bone coalition is common. Conducive hearing loss is seen and is due to fusion of the stapes to the petrous part of the temporal bone.
Defects in GDF5 are the cause of multiple synostoses syndrome type 2 (SYNS2) [MIM:610017]. Multiple synostoses syndrome is an autosomal dominant condition characterized by progressive joint fusions of the fingers, wrists, ankles and cervical spine, characteristic facies and progressive conductive deafness.
Defects in GDF5 are a cause of brachydactyly type A2 (BDA2) [MIM:112600]. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits.
Genetic variations in GDF5 are associated with susceptibility to osteoarthritis type 5 (OS5) [MIM:612400]. Osteoarthritis is a degenerative disease of the joints characterized by degradation of the hyaline articular cartilage and remodeling of the subchondral bone with sclerosis. Clinical symptoms include pain and joint stiffness often leading to significant disability and joint replacement.
Defects in GDF5 may be a cause of brachydactyly type A1 (BDA1) [MIM:112500]. Brachydactylies (BDs) are a group of inherited malformations characterized by shortening of the digits due to abnormal development of the phalanges and/or the metacarpals. They have been classified on an anatomic and genetic basis into five groups, A to E, including three subgroups (A1 to A3) that usually manifest as autosomal dominant traits.
Similarity:
Belongs to the TGF-beta family.
SWISS:
P43026
Gene ID:
8200
Database links:Entrez Gene: 8200 Human
Entrez Gene: 14563 Mouse
Omim: 601146 Human
SwissProt: P43026 Human
SwissProt: P43027 Mouse
Unigene: 1573 Human
Unigene: 4744 Mouse
Important Note:
This product as supplied is intended for research use only, not for use in human, therapeutic or diagnostic applications.
- 产品图片
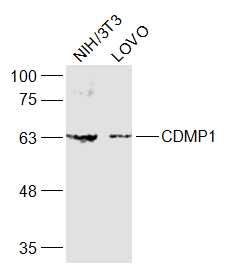 Sample:
Sample:
NIH/3T3(Mouse) Cell Lysate at 30 ug
LOVO(Human) Cell Lysate at 30 ug
Primary: Anti-CDMP1 (bs-6580R) at 1/1000 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 55 kD
Observed band size: 60 kD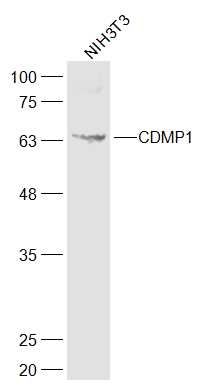 Sample:
Sample:
NIH/3T3(Mouse) Cell Lysate at 30 ug
Primary: Anti-CDMP1 (bs-6580R) at 1/300 dilution
Secondary: IRDye800CW Goat Anti-Rabbit IgG at 1/20000 dilution
Predicted band size: 55 kD
Observed band size: 60 kD